8. NiO: Hartree-Fock case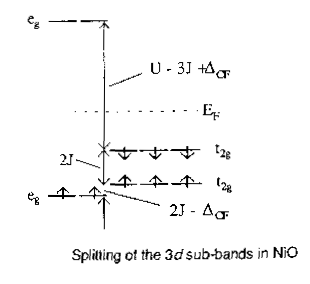 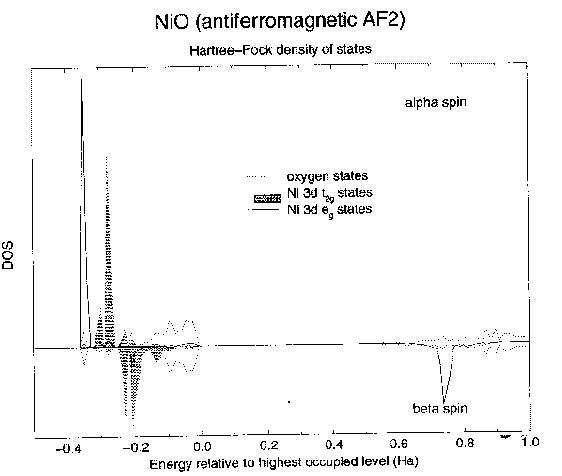 |



|
So, let's start by looking at the results of a simple Hartree-Fock calculation for NiO in the experimentally observed AF2 antiferromagnetic state (i.e. with up and down spins arrayed as in the picture on the earlier overhead). Above is a plot of the calculated density of states. I'll explain how I plotted the picture. What I've done is to arbitrarily multiply the minority spin density of states by -1 just to flip it over the energy axis so we can see it. The upper panel therefore corresponds to spin up, the lower panel to spin down. Now, at each energy we're just integrating over the Brillouin zone and counting states with that energy and I can project out the contributions from different orbitals. You can see the total oxygen, the Ni t2g and Ni eg as dotted line, shaded, and continuous line peaks respectively. Interestingly, the ordering of the states is the same as in the toy picture reproduced at the top of the overhead, with the added bonus of band width and all the intersite interactions. So we have two aspects to the picture. One, this is an insulator. In fact, it's a charge transfer insulator: the oxygen states are, correctly, the states just below the Fermi level. Two, we have essentially the correct Mott Hubbard physics. Because this is Hartree-Fock, the eigenvalues mean something (Koopmans' theorem). The lowest unoccupied states within a frozen orbital picture correspond roughly to the energy of a current carrying excitation, because the unoccupied states feel a d8 potential and the occupied states feel a d7 potential. This is the kind of charge fluctuation that appears in Mott's theory. This gap does not correspond to a local ('promotion') excitation. Now the third aspect: is the ground state an 'AF2' antiferromagnet with opposite spin planes going up the 111 direction? To do that we need to converge solutions in different magnetic states and look at the difference in energy between them. In general, to start off the iterations in an SCF calculation you need to have a good initial guess of what the density might be, so what the code I'm using does is to solve the atomic Hartree-Fock equations in order to get a spherical atomic density for each type of atom. You then stick these at the relevant points in the crystal structure and start with a superposition of atomic densities as your initial guess. The whole self-consistent field procedure then involves modifying the density to take account of the new crystalline environment, forming bonds, magnetic superexchange interactions and so on.. Now the HF atomic solution for Ni2+ is spin polarized, so obviously you just need to multiply the initial spin density matrix of the atoms by -1 to flip the spins for selected ions in the system and it will generally converge to the solution that you want and you can compare the energetics.. So we can flip any spin we like and play with different magnetic states. The AF2 state is correctly predicted to be the lowest in energy in preference to a ferromagnetic state or other possible antiferromagnetic states. |