Mike
Towler
Theory of Condensed Matter
Cavendish Laboratory, University of Cambridge
Disclaimer: the authors of the CRYSTAL program are not
responsible for the content of this site.
Please note that this page isn't really maintained any more,
since I no longer
work much with the CRYSTAL program.
|
CRYSTAL is a commercially available quantum mechanical electronic structure package which is able to calculate the electronic structure both of molecules and of systems with periodic boundary conditions in 1, 2 or 3 dimensions (polymers, slabs and crystals) using either Hartree-Fock or density functional theory. The program was written by Roberto Dovesi, Vic Saunders, Carla Roetti, Mauro Causà, Nic Harrison, Roberto Orlando and Claudio Zicovich-Wilson. The latest version is CRYSTAL2017 - this page refers largely to CRYSTAL98 and earlier versions - if I have time one day I might update it to reflect current realities (note that some of the utilities and stuff here have been updated to the latest 2017 version). I spent a couple of years in Torino as a postdoc in Roberto Dovesi's group (1994-1996), hence my interest. Although currently the main research effort of our group here in the Cavendish Laboratory is devoted to the development of quantum Monte Carlo methods with the CASINO program, CRYSTAL is often used in the generation of trial wave functions for this code. This page is therefore intended to collect together various useful resources for the program for the benefit of members of the Cambridge Theory of Condensed Matter (TCM) group. I hope it will also be of use to other members of the CRYSTAL user community. Any feedback would be appreciated.
CRYSTAL uses a local basis set of Gaussian-type functions to construct the sets of Bloch functions in which the one-electron crystalline orbitals are expanded. However unlike, say, a plane wave basis, Gaussian functions do not constitute a universal set, and individual basis sets have to be developed for each atom in the periodic table. Optimal Gaussian basis sets for use in periodic Hartree Fock/DFT calculations are often significantly different to those appropriate for molecular studies. The main difference is that in close packed solids one must be careful to avoid the problems of linear dependence and basis set superposition error due to the overlap of very diffuse functions. In molecular basis sets these functions are necessary in order to describe the long range decay of the wave function and are thus not required in the solid. An article I wrote for a summer school in Torino a long time ago (and now updated for a more recent school) discussing the use of Gaussian basis sets for periodic calculations is here. It's hardly a work of significant scholarship but it should serve as a useful introduction. The following table will access a new improved CRYSTAL Gaussian basis set library. The basis sets provided here have largely been developed and used by the Daresbury group, the Torino group and me. Clicking on any element should reveal a text file containing appropriate basis sets; often there will be several alternatives which may be more or less suitable for any given problem. An attempt has been made to attribute authorship to each basis set and to provide references to their use in the literature. The database is of course far from complete, and any additional contributions would be welcome (mail me : mdt26 at cantab.net) . NB: Many of the basis sets provided here (mainly those beyond Zn) have never been used, having simply been optimized via atomic Hartree-Fock calculations. They should therefore be considered only as a starting point for more serious work. The files generally contain comparisons with published HF limit energies and suggestions for how to optimize the valence functions for solid-state calculations. I would be delighted to hear from anybody who uses or optimizes these basis sets in real calculations. NB2: (16/7/99) This is not a copy of the recently published basis set library on the Torino CRYSTAL site. Both libraries have their origin in a set of files I compiled in 1995 whilst working in Torino. However, their content has diverged over the last 5 years and you may find it useful to check both sites when looking for suitable basis sets for given problem. Note that I am also more liberal than the Torino people in allowing basis sets which have not been used in publications, hence the presence of the large collection of never-used post-Zn elements. |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Here is a table of Hartree-Fock solutions for atomic ground states (Z=2-54) near the Hartree-Fock limit (due to Bunge, Barrientos and Bunge). You may find this useful for comparative purposes when optimizing your own Gaussian basis sets (the billy program described in my summer school article and available for download below will do this for you). A useful database of molecular Gaussian basis sets is here. With appropriate adaptations these can be useful starting points for solid calculations.
I have collected together a reasonably complete list of articles referencing the CRYSTAL program written by absolutely anyone from 1981-2000. If you want to know whether anyone has investigated plutonium-doped lanthanum nickelate (or whatever) before, this is where you find out. Surprisingly, nobody has. (Note that the official sites now do something similar to this - so you may find extra references there..)
(1) How to optimize Gaussian basis sets for CRYSTAL calculations.
Here is a list of references to publications involving use or development of the CRYSTAL program involving members of the TCM group:
Interface to quantum Monte Carlo code (Mike Towler) The quantum Monte Carlo method and our implementation of it for crystalline
solids is discussed in detail on our quantum Monte
Carlo page. Strongly correlated systems (Mike Towler/Richard Needs/Nic Harrison) A question of interest for many years has been whether one can describe so-called 'strongly correlated' systems with first principles electronic structure calculations. While in simple metals, semiconductors and many ionic materials first principles calculations now underpin our understanding of the electronic structure and bonding, attempts to apply first principles methods to strongly correlated systems such as NiO and the high Tc cuprates have been fraught with difficulties. Until recently almost all calculations were based on the independent electron approximation as embodied in the local density approximation (LDA) to density functional theory (DFT). When the LDA is used to describe the magnetically ordered, insulating ground states of materials such as La2CuO4 a non-magnetic, metallic ground state is obtained (see eg: Wilson E Pickett Rev. Mod. Phys. 61 433 1989). For many years such results were thought to be a failure of the independent electron approximation per se and proof positive that such highly correlated systems could not be described using band theory. It is now apparent that this is not the case. The failure of the LDA was due to its approximate treatment of the exchange interaction. Indeed a variety of approaches (LDA+U, SIC-LDA) have now been developed to introduce better descriptions of the on-site exchange interaction. Beginning in 1993 a collaboration between the Daresbury and Torino groups (also including myself, Bill Mackrodt of St. Andrews University and Neil Allan of Bristol University) was initiated with the intention of developing a different approach to these systems. The key to this approach has been the realisation that the magnetically ordered insulating ground state may often be described by a single determinant wave function and may therefore be accurately represented using the Hartree-Fock approximation which, by definition, contains the exact exchange interaction. The performance of such calculations and hybrid schemes using combinations of DFT and exact exchange has now been well documented in a variety of publications. Here is a page giving references to all publications arising out of this collaboration since 1993 (including those by other groups who have since begun to use this method). Here in Cambridge it is now our intention to use CRYSTAL trial wave functions for highly accurate quantum Monte Carlo calculations of strongly correlated materials, focussing initially on the 3d transition metal monoxides. This project is expected to begin over the next few months. This link will take you to the text and overheads of a talk that I presented recently (25th Feb 1998) entitled 'Strongly Correlated Materials in Electronic Structure Theory'. This was a supplementary lecture given as part of Professor Peter Littlewood's graduate course on correlated electron physics, which I gave with the (completely unsuccesful) aim of provoking discussion with the many-body theory community. An excellent recent summary of what can be done with CRYSTAL for strongly correlated magnetic insulators is Nic Harrison's March 1998 paper: 'Transition metal materials: a first principles approach to the electronic structure of the insulating phase' Phil. Trans. Roy. Soc. London, 356, 75-87 (1998). Muonium as a hydrogen analogue in silicon and germanium; quantum effects and hyperfine parameters (Andrew Porter/Mike Towler/Richard Needs) Hydrogen is an important and very common impurity in semiconductors such as silicon. Its presence can dramatically alter the electrical and optical properties of such materials and, since it is present in many of the stages in their manufacture, it is very hard to be free of it.
The aim of the project is to use CRYSTAL to study the hyperfine tensor of the muon at the sites believed to be favoured in crystalline silicon. Since the muon is considerably lighter than the proton (with only one ninth its mass), it has a far larger zero point energy. By establishing the shape of the potential well that the muon experiences and solving the Schrodinger equation for the muon in this well, we can calculate the size of this zero point energy and study its effect on the overall stability of the site. [PDF file available here (Phys. Rev. B 60, 13534 (1999))] Carbon clusters near the crossover to fullerene stability. (Paul Kent/Mike Towler/Richard Needs/Guna Rajagopal) Since the discovery of fullerene C60 in the 1980s, the mechanisms of fullerene formation have been an area of considerable research. Considerable attention has been devoted to the question ``which is the smallest stable fullerene?''. While fullerenes are not seen experimentally for clusters smaller than C28, many theoretical studies suggest that smaller clusters may have a fullerene ground state. Previous calculations of C20, the smallest carbon cluster that could in principle exist as a fullerene cage, showed that an accurate treatment of electron correlation was crucial, due to the subtle interplay of Coulomb, exchange and correlation energies. QMC calculations by Mitas and Grossman have showed that a bowl-shaped structure is the lowest energy C20 isomer. In this investigation, we apply fixed-node diffusion Monte Carlo (DMC) to accurately compute the ground state total energies of various structures of carbon clusters with 24, 26, 28 and 32 atoms. We also compare the Quantum Monte Carlo results with those of CRYSTAL95 DFT calculations using a range of different functionals. [PDF file available here (Phys. Rev. B 62, 15394 (2000)] Electronic and magnetic structure of RuSr2GdCu2O8. (Mike Towler)
More details soon.
Any questions about the functionality of the program, how to obtain the code or other similar queries should be directed to the Torino group. Any questions about issues raised on this page can be mailed to me ( mdt26 at cantab.net).
|
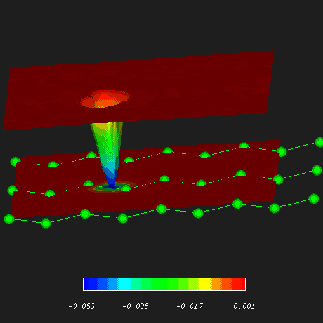 In
QMC calculations one generally starts from a
a set of one-electron orbitals generated with a density functional
or Hartree-Fock code, the determinant of which (times a correlating Jastrow factor)
is used as a trial wave function for a QMC run. Until now we have only been able to use an
in-house plane-wave DFT package (K207 or 'out-of-date-TEP') to compute
the trial wave function; I have recently rewritten our QMC code to
give us the additional option of running the Monte Carlo in a local
basis of Gaussian functions taking input from the CRYSTAL program. I have
added additional functionality to the QMC code to allow us to compute excited
states (by switching selected occupied and virtual orbitals in the
CRYSTAL one-electron determinant) and to use multi-determinantal trial
wave functions. Preliminary results are very good. As an example, I am
currently using diffusion Monte Carlo to calculate the excitation spectrum
of diamond - the principal band gap is found to be 7.2 eV (+-0.1eV) compared
with the experimental result of 7.3 eV [click
In
QMC calculations one generally starts from a
a set of one-electron orbitals generated with a density functional
or Hartree-Fock code, the determinant of which (times a correlating Jastrow factor)
is used as a trial wave function for a QMC run. Until now we have only been able to use an
in-house plane-wave DFT package (K207 or 'out-of-date-TEP') to compute
the trial wave function; I have recently rewritten our QMC code to
give us the additional option of running the Monte Carlo in a local
basis of Gaussian functions taking input from the CRYSTAL program. I have
added additional functionality to the QMC code to allow us to compute excited
states (by switching selected occupied and virtual orbitals in the
CRYSTAL one-electron determinant) and to use multi-determinantal trial
wave functions. Preliminary results are very good. As an example, I am
currently using diffusion Monte Carlo to calculate the excitation spectrum
of diamond - the principal band gap is found to be 7.2 eV (+-0.1eV) compared
with the experimental result of 7.3 eV [click  Unfortunately,
it is difficult to study hydrogen in situ in the materials of interest
because it has only a weak paramagnetic interaction. As a consequence,
muons having the same charge as the proton but a far larger nuclear magnetic
moment are used to mimic hydrogen in experiments to study the stable locations
of the impurity within the crystal lattice. These favoured sites can be
characterized by the hyperfine interactions of the muon.
Unfortunately,
it is difficult to study hydrogen in situ in the materials of interest
because it has only a weak paramagnetic interaction. As a consequence,
muons having the same charge as the proton but a far larger nuclear magnetic
moment are used to mimic hydrogen in experiments to study the stable locations
of the impurity within the crystal lattice. These favoured sites can be
characterized by the hyperfine interactions of the muon.