CASINO works as stand-alone software only for certain model systems like the homogeneous electron gas. For real systems containing atoms it requires input from another program – such as a Hartree-Fock, DFT, or quantum chemistry program – to define a ‘trial wave function’ which the DMC process is then supposed to improve. Hence QMC ‘makes the answer better’. Unluckily for the CASINO developers, quantum Monte Carlo codes therefore require the development and long-term support of standard interfaces with multiple external programs.
For the case of CASINO, an external code needs to write an ‘xwfn.data‘ file, where x is a letter indicating the kind of basis set (p=plane-wave, b=blip, g=gaussian, sto=slater, a=numerical atomic orbitals on a grid, d=numerical molecular orbitals on a grid for diatomics). These files contain (1) some basic information about the system and the generating program, (2) the geometry, (3) the basis set, (4) any multideterminant information, and (4) the orbital coefficients (the numbers that multiply the basis functions when you’re linearly combining them to represent an orbital). For the awfn.data and dwfn.data cases, (3) and (4) are not present, and the value of the orbital is given directly on some kind of grid. The exact format of these files is defined in a formal specification in Section 7 of the CASINO user manual. Anyone who has some knowledge of the internal workings of a third-party code, or is in possession of a proper definition of the output of such a code, should find it easy enough to develop a simple converter program to write a CASINO xwfn.data file and thus construct a formally defined interface. This can take the form of an external utility which reads the output of the third-party code, or one can modify the third-party code to write the CASINO file directly. Potential QMC users are warmly encouraged to do so! More advice is given on this site here.
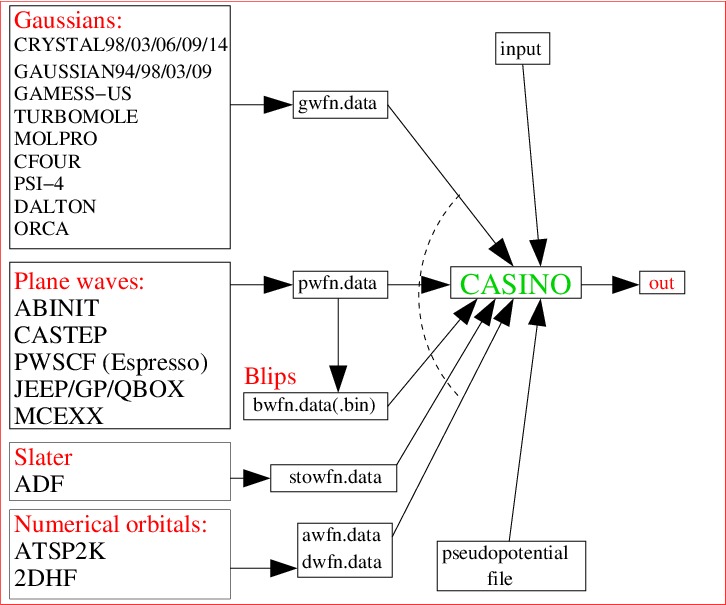
This page provides a list of third-party codes that – at least in principle – have support for producing CASINO trial wave function xwfn.data files. The exact degree of support will depend on time; in particular the interfaces may stop working if the developers release a new version of their code without bothering to check that CASINO support still works. If you find this is the case, please let us know. All voluntary contributions to maintaining these interfaces is gratefully accepted, since doing so is very boring indeed. Our personal support for the codes can be greatly enhanced if we have someone working in Cambridge who is a dedicated user of the code in question. When they leave, our group knowledge may become non-existent (e.g., currently, none of us know anything about the popular GAUSSIAN code).
Example input files for some of the codes below can be found in the distribution directory CASINO/examples/wfn_generation and in CASINO/examples/generic/gauss_dfg. Instructions are in accompanying READMEs.
Inside each of the basis set headers, the codes are given in a rough order of preference. The codes near the top of each list are likely to be widely used with a properly supported and maintained interface, and/or support more features.
Plane-wave/blip basis set
Although CASINO is capable of directly evaluating orbitals expanded in plane-waves (in its initial version, that was all it could do) this is now considered extremely inadvisable, as it adds an extra power of N to the scaling of the required computer time with system size. This is because every plane-wave potentially contributes at every point in space, and the number of necessary plane-waves increases with system size. It is far better to expand the orbitals in a localized basis set; then only a fixed number of basis functions can potentially contribute at any random point, and this number does not increase with system size. For users of plane-wave codes, the plane-wave orbitals can be re-expanded in a localized basis of blip functions using a well-defined mathematical transformation. PWSCF is currently the only code that can do this natively; the pwfn.data output of all other plane-wave codes will need to be transformed into a bwfn.data blip file by the CASINO utility program ‘blip‘.
(1) PWSCF (part of the Quantum Espresso package)
![]()
This is the best-supported plane-wave DFT package with native CASINO support and it’s free (amongst the developers and their friends it is used by Mike Towler and Dario Alfè). The CASINO support has three unique features: (1) PWSCF can do blip transformations natively and write out either pwfn.data or bwfn.data files (the latter in a binary format if requested), (2) the CASINO distribution contains a ‘runpwscf‘ run script that supports the CASINO architecture system, and (3) it supports DFT-DMC molecular dynamics calculations. CASINO also supplies a ‘twistav_pwscf‘ script which automates the process of performing twist-averaged QMC calculations.
Assuming you use the supplied ‘runpwscf‘ script, then one can generate an xwfn.data file simply by typing ‘runpwscf --qmc‘ (where runpwscf also supports all the usual runqmc command-line arguments specifying e.g. the number of cores etc. on any machine supported by CASINO, that is, one that has a defined CASINO_ARCH file).
Pseudopotential converters between PWSCF’s UPF format and CASINO’s x_pp.data format are supplied with PWSCF.
For more detailed information, see Section 8.10 of the CASINO manual.
(2) CASTEP
![]()
CASTEP is a powerful plane-wave DFT package that – like CASINO – was originally developed in the Cavendish Laboratory Theory of Condensed Matter Group. It is currently distributed by Accelrys as part of its Materials Studio package: see here. It is freely available to UK academics: see here . Mail Chris Pickard at [email protected] to discuss CASTEP and how to get hold of a copy. Of the main CASINO developers, Neil Drummond is the one who uses CASTEP as his principal DFT code.
You can generate a CASINO pwfn.data file by running the castep2casino utility (supplied with CASTEP) in a directory containing CASTEP output. This can be transformed into a blip representation using the CASINO-supplied blip utility.
CASINO’s x_pp.data pseudopotential format is supported natively by CASTEP.
For more detailed information, see Section 8.4 of the CASINO manual.
(3) ABINIT

Official ABINIT blurb:
“ABINIT is a package whose main program allows one to find the total energy, charge density and electronic structure of systems made of electrons and nuclei (molecules and periodic solids) within Density Functional Theory (DFT), using pseudopotentials and a planewave or wavelet basis. ABINIT also includes options to optimize the geometry according to the DFT forces and stresses, or to perform molecular dynamics simulations using these forces, or to generate dynamical matrices, Born effective charges, and dielectric tensors, based on Density-Functional Perturbation Theory, and many more properties. Excited states can be computed within the Many-Body Perturbation Theory (the GW approximation and the Bethe-Salpeter equation), and Time-Dependent Density Functional Theory (for molecules).”
ABINIT has native CASINO support. To tell ABINIT to write the wave function in CASINO format, set prtwf to 2 in the ABINIT input file. Some other input parameters must also be set appropriately.
Appropriate pseudopotential converters are provided in the CASINO distribution.
For more detailed information, see Section 8.1 of the CASINO manual.
(4) MCEXX
MCEXX is a code written by A. Görling, S. Rohra, P. Carrier, A. Hesselmann, H. Schulz and E. Trushin at the University Erlangen Nuremberg in Germany. It is a plane wave electronic structure code for conventional Kohn-Sham calculations (LDA/GGAs), Hartree-Fock calculations, DFT calculations with hybrid functionals, exact-exchange (EXX) Kohn-Sham calculations, and calculations treating electron correlation within the random phase approximation. Spin-orbit interactions, non-collinear spin, and accompanying magnetization currents can be treated. With explicitly temperature-dependent functionals calculations for the the very high temperatures relevant in warm dense matter physics (e.g. in the context of nuclear fusion or matter in stars) can be carried out. An interface between MCEXX and CASINO was implemented in November 2013. The code is not formally publically available, and no information has been provided on how to use the interface, but interested parties may contact Prof. Görling at [email protected].
(5) GP / JEEP / QBOX
We used to support an old Lawrence Livermore code which, when we played with it years ago, was called GP. It also appears to have been called JEEP, and has now morphed into something called QBOX which has a website here.
There is a converter called ‘jeep_to_pwfn‘ supplied with CASINO which used to read in GP ‘jeep.wf‘ and ‘atoms.sys‘ files to produce a CASINO pwfn.data file. It is highly unlikely that this still works with modern versions of QBOX. If anyone out there would like to update this information, or to make the converter work in a modern context, then they would be very welcome.
Gaussian basis set
(6) CRYSTAL (95/98/03/06/09/14/17)

CRYSTAL is a commercially available Hartree-Fock/DFT Gaussian basis set package which is able to treat both molecules and systems with periodic boundary conditions in 1, 2 or 3 dimensions (polymers, slabs or crystals). It originated at the University of Torino in Italy, with considerable collaboration from Daresbury Laboratory and other institutions in the UK. The author list of the latest version of the program is Roberto Dovesi, Vic Saunders, Carla Roetti, Roberto Orlando, Claudio Zicovich-Wilson, F. Pascale, Bartolomeo Civalleri, Klaus Doll, Nic Harrison, Ian Bush, Philippe D’Arco, Miquel Llunell, Mauro Causa, Y. Noel, L. Maschio, A. Erba, M. Rerat and Silvia Casassa.
CRYSTAL has the best CASINO support of all the available Gaussian basis codes, and has the unique advantage of treating periodic systems (which is done either badly or – more usually – not at all in other Gaussian codes that we know about). Versions of CRYSTAL are supported back to CRYSTAL95, but we strongly recommend that you use the latest CRYSTAL17 edition.
THE CASINO distribution includes a ‘runcrystal‘ shell script which will produce a gwfn.data file if invoked with the -qmc command line option (note this is currently still an old script which does not use the CASINO architecture system, and it may therefore need some manual setup). Without this script the procedure for generating the gwfn.data file is somewhat more involved; full details are provided in Section 8.4. of the CASINO manual.
Trail-Needs pseudopotentials (and some corresponding Gaussian basis sets) are provided in our pseudopotential library in CRYSTAL format. Other basis sets and various other somewhat out-of-date material can be found on Mike Towler’s old CRYSTAL website – which he made because his first job after getting his Ph.D. involved spending two years in Torino in Roberto Dovesi’s group (Mike should therefore be the first person you ask about any issues about the CASINO-CRYSTAL interface).
(7) GAUSSIAN (94/98/03/09)

GAUSSIAN is an extremely large and widely used commercially available quantum chemistry package which implements pretty much every quantum chemistry technique. That said, the CASINO distribution contains the utility ‘gaussiantoqmc‘ which can read in GAUSSIAN formatted checkpoint files (along with the standard output file) and produce a CASINO gwfn.data.
The GAUSSIAN-CASINO interface can supposedly deal with the following sorts of calculation:
- Hartree-Fock and DFT ground states, open and closed shell.
- CIS excited states, open and closed shell.
- CASSCF states.
- Time-dependent HF (TD-HF) or DFT (TD-DFT) excited states.
A large amount of detailed information about how to use gaussiantoqmc is given in Section 8.6 of the CASINO manual.
Trail-Needs pseudopotentials (and some corresponding Gaussian basis sets) are provided in our pseudopotential library in GAUSSIAN format.
Note that if the gaussiantoqmc utility doesn’t do what you want (and it may not, since it’s 15 years old, and the person who wrote it disappeared not long afterwards) then you might ask Katharina Doblhoff-Dier, who has apparently written a new equivalent utility from scratch (ask MDT for her contact details if you can’t find her on the internet).
(8) GAMESS-US
“GAMESS is a program for ab initio molecular quantum chemistry. Briefly, GAMESS can compute SCF wave functions ranging from RHF, ROHF, UHF, GVB, and MCSCF. Correlation corrections to these SCF wave functions include Configuration Interaction, second order perturbation Theory, and Coupled-Cluster approaches, as well as the Density Functional Theory approximation. Excited states can be computed by CI, EOM, or TD-DFT procedures“.
There are two alternatives for the CASINO interface to the GAMESS-US package:
- An old set of Perl scripts distributed with the main CASINO distribution, the main one of which is called ‘
gamess2qmc‘. These live, along with aREADMEfile with instructions and some basic examples, in the directoryCASINO/utils/wfn_converters/gamess.They were originally written by Alexander Badinski many years ago. Support for f functions was added in 2015 by Kevin Gasperich. The script has considerable limitations and some users have reported errors. A typical comment from an (understandably) anonymous user was: “However, I admit that the gamess2qmc converter is rubbish! It is very unstable for different GAMESS versions and leads to basically unpredicted results with NO error catches. I would strongly discourage anyone from using it without carefully adapting it by hand to the GAMESS version used.” Make of that what you will, but providing you have some idea of what you’re doing and you’re not doing anything weird it is not so difficult to make it work. - (2) Albert Defusco of the University of Pittsburgh has implemented native CASINO support into GAMESS independently of the
gamess2qmcscript. This is certainly available in the current standard GAMESS distribution; the facility has been evolving and it might be best to say you should use the most recent version available (I – MDT – have been made aware [July 2015] of a problem occuring when basis functions are automatically discarded because of quasi-linear dependence. . last I heard a fix was being worked on..).
(9) TURBOMOLE
![]()
TURBOMOLE is a quantum chemical program package, initially developed in the group of Prof. Dr. Reinhart Ahlrichs at the University of Karlsruhe and at the Forschungszentrum Karlsruhe, and now run by a commercial company. It provides all standard and state of the art methods for ground state calculations (Hartree-Fock, DFT, MP2, CCSD(T)), excited state calculations at different levels (full RPA, TDDFT, CIS(D), CC2, ADC(2), …), geometry optimizations, transition state searches, and molecular dynamics calculations.
The CASINO converter/interface was implemented a long time ago (2001) by Stefan Grimme of the University of Muenster, and has almost certainly stopped working. A piece of code which was to be incorporated into the TURBOMOLE source is provided in CASINO/utils/wfn_converters/turbomole_old. (Martin Korth of the University of Ulm was the last person known to possibly have his own private version which works).
In modern times the interface to Turbomole has in fact been revived through its ability to write out MOLDEN files. These can be read by the molden2qmc converter (written by Mike Deible and Vladimir Konjkov) which is included in the current CASINO distribution.
(10-13) MOLPRO , CFOUR, PSI4, DALTON, ORCA (NWCHEM, Q-CHEM..)
![]()

Mike Deible and Vladimir Konjkov have written a converter molden2qmc that will write a gwfn.data file from information written in the standard MOLDEN file format, with the intention of supporting all five of the codes above. Vladimir has also indicated to me that it may be possible to add NWCHEM and Q-CHEM to this list.
According to Google, one finds that the MOLDEN format is also supposedly supported in some sense by ACESII, MOLCAS, Jaguar, CADPAC, GEOMOP, and HONDO (whatever they might be) thus it may ultimately be possible to provide some sort of connection with these codes too.
Support is now in the CASINO distribution for all five of these codes. Some additional notes regarding the idiosyncrasies of particular codes can be found in the relevant section of the manual.
Note in particular that CFOUR’s implementation of MOLDEN is a little special.. and requires various vectors to be reordered before the standard CASINO molden2qmc converter will work. To do this, replace the file /libr/reorderdf.f in the CFOUR distribution with the file utils/wfn_converters/cfour/reorderdf.f from the CASINO distribution, then recompile CFOUR.

Slater basis set
(13) ADF
ADF is a DFT code that uses Slater basis sets.
The CASINO converter adf2stowf.py – which is a Python script (whatever that might be) – was developed for version 2008.01 of the ADF code by a TCM postdoc (Norbert Nemec) who has since left. It takes the output of a molecular ADF calculation and produces a CASINO trial wave function file (stowfn.data) in a Slater-type basis.
The ADF converter and the Slater-type orbital implementation in CASINO have not yet been properly tested, so expect bugs and limitations, which will need to be fixed by a competent person who understands what on earth is going on.
Further useful information may be available in Section 8.2 of the CASINO manual, and in the README file in CASINO/utils/wfn_converters/adf .
Numerical orbitals
CASINO is capable of using numerical orbitals represented on grids for atoms and diatomic molecules.
(14) ATSP2K
For the atomic case we supply a converter ‘extractdet‘ which converts the output of the program A2SP2K into CASINO awfn.data format (further details are in section 8.3 of the CASINO manual).
(15) 2DHF
For the diatomic case, one may use the code 2DHF. Its output may be converted into the CASINO dwfn.data format using the supplied CASINO utility ‘uf2dwfn‘. Further details are in section 8.12 of the CASINO manual.
Both utilities were written by John Trail, and he (jrt32 at cam.ac.uk) should be your first point of contact for numerical atomic or diatomic issues.